# Change this line appropriately
from answers.Bioimage_analysis_05_answers_bacteria.bacterial_seg_functions import *
# Put the location of the data here.
path_ecoli_data = "/Users/m.wehrens/Data_notbacked/2025_Py-Image-workshop_Filamentation-example-data/pos3crop_timelapse_switch890.98.tif"
# Load data
imgs_ecoli = tiff.imread(path_ecoli_data)Workshop Python Image Analysis
Martijn Wehrens, May 2026
The code below allows you to segment bacteria.
It has some additional features compared to what was discussed during previous workshop chapters. It is still not perfect, because this is a very tricky dataset to segment. For the related paper, many manual corrections where performed.
You can play around with this code to see what it does, e.g. go through the code line-by-line and check what different parts do and how parameters in the code look.
The contents of the file bacterial_seg_functions.py are printed below, copy and paste these to a local bacterial_seg_functions.py on your computer.
"""
Functions for bacterial segmentation.
"""
################################################################################
# %% Libraries
import matplotlib.pyplot as plt
from matplotlib.colors import ListedColormap
import seaborn as sns
import tifffile as tiff
import numpy as np
import skimage as sk
from skimage.util import invert
from scipy import stats
from scipy import ndimage
from skimage.measure import label, regionprops
################################################################################
# %% Helper functions
def cmap_random(N):
"""Create a cmap with 500 random ±bright colors, starting with black"""
cmap = np.zeros((N+1, 3))
for i in range(1, N+1):
cmap[i] = np.random.rand(3)*0.7 + 0.3
return ListedColormap(cmap)
def _global_mask(input_img, disksize_range=20):
""" Determine a global mask based on local intensity differences.
Assuming the area around the bacteria has more intensity differences
than the background, we can identify the general area where the
bacteria are.
"""
# input_img = invert(imgs_ecoli[10,:,:])
# determine local difference ranges
# - "img_delta" will contain the difference between the local maxima and local
# minima around each respective pixel.
img_min = ndimage.minimum_filter(input_img, footprint=sk.morphology.disk(disksize_range))
img_max = ndimage.maximum_filter(input_img, footprint=sk.morphology.disk(disksize_range))
img_delta = img_max - img_min
# plt.imshow(img_delta)
# Use an otsu threshold to select regions with high local differences (i.e. bacteria)
thresh = sk.filters.threshold_otsu(img_delta)
mask_global = img_delta > thresh
# plt.imshow(mask_global)
# Fill holes
# - Above works, but resulting mask has holes. Fill those.
mask_global_fill = ndimage.binary_fill_holes(mask_global)
# Select largest region (there might be other small artifact regions)
labeled_global = sk.measure.label(mask_global_fill)
largest_region = np.argmax([r.area for r in sk.measure.regionprops(labeled_global)]) + 1
mask_global_filled = labeled_global == largest_region
# plt.imshow(mask_global_filled)
# Now use erosion, as the mask is oversize due to neighborhood size
border_margin=5 # keep a certain margin still, though
disksize_shrink = np.max([0, disksize_range-border_margin])
mask_global_final = \
sk.morphology.erosion(
mask_global_filled,
footprint=sk.morphology.disk(disksize_shrink)
)
# plt.imshow(mask_global_final)
# Plot the result (debugging only)
# plt.imshow(input_img); plt.contour(mask_global_final, levels=[0.5], colors='r')
# now also obtain related binding box
bbox = sk.measure.regionprops(mask_global_final.astype(int))[0].bbox
return mask_global_final, bbox
def _get_edges(input_img_crop, sigma_smooth, disksize_crossing, showplot=False):
""" Find edges in an image using Laplacian of Gaussian (LoG) appraoch. """
# Smooth image before Laplacian filtering
img_gauss = sk.filters.gaussian(input_img_crop, sigma=sigma_smooth)
# Apply laplacian
img_laplacian = sk.filters.laplace(img_gauss)
if showplot:
minmaxval = np.min([-np.min(img_laplacian), np.max(img_laplacian)])
plt.imshow(img_laplacian, cmap='bwr', vmin=-minmaxval, vmax=minmaxval)
plt.show()
# plt.hist(img_laplacian.ravel(), bins=100)
# Identify LoG zero crossings as bacterial outlines
# A pixel is considered crossing zero if in its neighborhood there are
# both negative and positive pixels.
# So search neighborhood for minimal and maximum values
edges_min = ndimage.minimum_filter(img_laplacian, footprint=sk.morphology.disk(disksize_crossing))
edges_max = ndimage.maximum_filter(img_laplacian, footprint=sk.morphology.disk(disksize_crossing))
# And check if this search yielded both negative and positive number
mask_edges = np.logical_and(edges_min < 0, edges_max > 0)
if showplot:
plt.imshow(mask_edges, cmap='gray')
return mask_edges
def _bacterial_seeds(input_img_crop, min_distance=5, showplot=False):
""" Identify bacterial locations using local maxima.
This simple function only aims to find locations that cover the insides
of all bacteria, but multiple locations per bacteria are OK.
(Identifiying individual bacteria will be done with different seeds.)
"""
# apply gaussian
img_smooth = sk.filters.gaussian(input_img_crop, sigma=3)
# get local maxima
seed_locations = sk.feature.peak_local_max(img_smooth, min_distance=min_distance)
# plot the result
if showplot:
plt.imshow(input_img_crop)
for x,y in seed_locations:
plt.plot(y,x,'ro')
plt.show()
return seed_locations
def _flood_fill_multiple(mask_edges, seed_locations):
""" Perform flood-fill per seed to get bacterial areas. """
# Create an empty mask to hold the filled regions
mask_filled = mask_edges.copy()
# Loop over each seed location and perform flood fill
for x, y in seed_locations:
# (x, y) = seed_locations[0]
mask_filled = \
sk.morphology.flood_fill(
mask_filled,
(x, y),
2)
# plt.imshow(mask_filled)
return mask_filled
def _unique_seeds_erosion(mask_bacteria, disksize_bacshrink=5):
""" Aims to obtain seeds that correspond to unique indivual bacteria.
Uses strategy based on erosion."""
# Use erosion on preliminary bacterial mask to get areas that each match
# one individual bacteria
mask_bacteria_shrunk = \
sk.morphology.erosion(mask_bacteria,
footprint=sk.morphology.disk(disksize_bacshrink))
return sk.measure.label(mask_bacteria_shrunk)
def _unique_seeds_distance(mask_bacteria, sigma_seed, distance_threshold):
""" Aims to obtain seeds that correspond to unique individual bacteria.
Uses strategy based on distance map.
Distance threshold should be similar but smaller than
the expected minimum width of the bacteria.
"""
# sigma_seed=3; distance_threshold=3
# create distance map
distance_map = ndimage.distance_transform_edt(mask_bacteria)
# median filter on distance map (filtering out local noise)
distance_map_blur = sk.filters.gaussian(distance_map, sigma=sigma_seed)
# plt.imshow(distance_map_blur, cmap='jet')
# keep areas with sufficient distance-to background to be inside
# the bacteria (similar to erosion)
mask_seeds = distance_map_blur > distance_threshold
# plt.imshow(mask_seeds)
return sk.measure.label(mask_seeds)
################################################################################
# %% Segmentation function
def seg_bacterium(input_img, sigma_smooth = 3,
disksize_crossing=1, disksize_range=20, min_distance=5,
sigma_seed=3, distance_threshold=3):
# sigma_seed=2, distance_threshold=4):
"""
Segment bacteria in an image using LoG zero crossings and watershed.
"""
# input_img = invert(imgs_ecoli[10,:,:])
# input_img = invert(imgs_ecoli[100,:,:])
# input_img = invert(imgs_ecoli[1000,:,:])
# plt.imshow(input_img)
# sigma_smooth = 3; disksize_crossing=1; disksize_range=20; min_distance=5; sigma_seed=2; distance_threshold=5
# identify part of the image with the colony
mask_global, bbox = _global_mask(input_img, disksize_range=disksize_range)
# plt.imshow(mask_global)
# .. and crop the image for speed
input_img_crop = input_img[bbox[0]:bbox[2], bbox[1]:bbox[3]]
mask_global_crop = mask_global[bbox[0]:bbox[2], bbox[1]:bbox[3]]
# plt.imshow(mask_global_crop)
# Get edges
mask_edges = _get_edges(
input_img_crop,
sigma_smooth = sigma_smooth,
disksize_crossing=disksize_crossing
)
# plt.imshow(mask_edges)
# Fill all the areas inside the edges where bacteria are suspected
seed_locations = _bacterial_seeds(input_img_crop, min_distance=min_distance)
mask_filled = _flood_fill_multiple(mask_edges, seed_locations)
# plt.imshow(mask_filled)
# Remove parts outside the global colony
mask_bacteria = np.logical_and(mask_filled, mask_global_crop)
# plt.imshow(mask_bacteria)
# Identify seeds matching individual bacteria
mask_unique_seeds = \
_unique_seeds_distance(mask_bacteria,
sigma_seed=sigma_seed,
distance_threshold=distance_threshold)
# plt.imshow(mask_unique_seeds)
# Now apply watershed to get individually labeled bacteria
result = sk.segmentation.watershed(
-1 * mask_bacteria,
markers=mask_unique_seeds,
mask=mask_bacteria)
# plt.imshow(result, cmap=)
# plt.imshow(result, cmap=cmap_random(200))
return result, input_img_crop
################################################################################ Example of how to use these functions:
# Show a few example images
plt.imshow(invert(imgs_ecoli[10,:,:]))
plt.show()
plt.imshow(invert(imgs_ecoli[300,:,:]))
plt.show()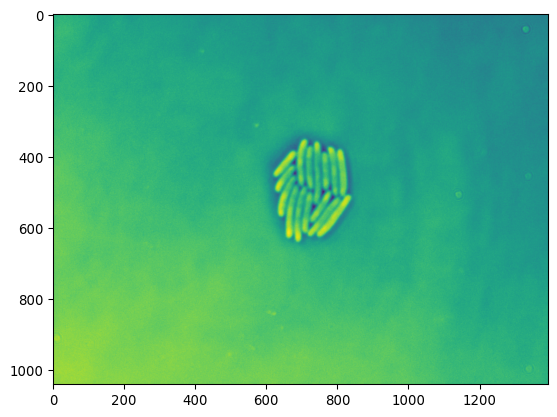
plt.imshow(invert(imgs_ecoli[1050,:,:]))
plt.show()
# Segment a single frame
segmask_oneframe, img_inv_crop = seg_bacterium(invert(imgs_ecoli[1000,:,:]))
_ = plt.imshow(segmask_oneframe, cmap=cmap_random(300))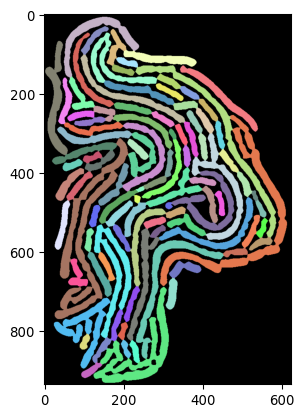
# Function to show segmentation result
def plot_img_and_segresult(img_inv, segmask):
"""Provided inverted original, plot image and result on top using contour"""
fig, axs = plt.subplots(1,2, figsize=(17/2.54,9/2.54))
axs[0].imshow(img_inv, cmap='gray')
axs[0].contour(segmask, levels=[0.5], colors='r', linewidths=.5)
axs[1].imshow(img_inv, cmap='gray')
axs[1].imshow(segmask, cmap=cmap_random(300))
plt.tight_layout()
plt.show()# Examples for segmentation results throughout the dataset
current_segmask, current_img_inv_crop = \
seg_bacterium(invert(imgs_ecoli[10,:,:]))
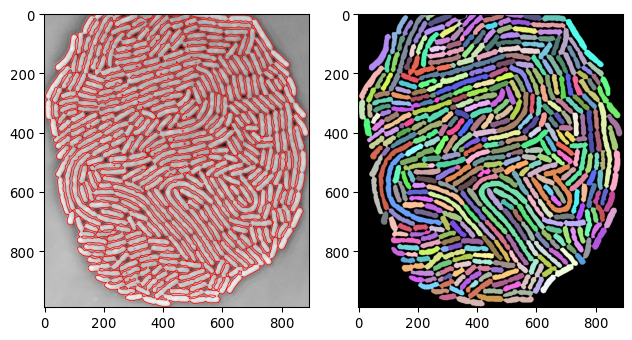
plot_img_and_segresult(current_img_inv_crop, current_segmask)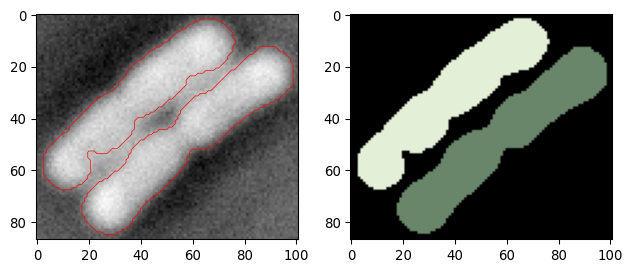
current_segmask, current_img_inv_crop = \
seg_bacterium(invert(imgs_ecoli[100,:,:]))
plot_img_and_segresult(current_img_inv_crop, current_segmask)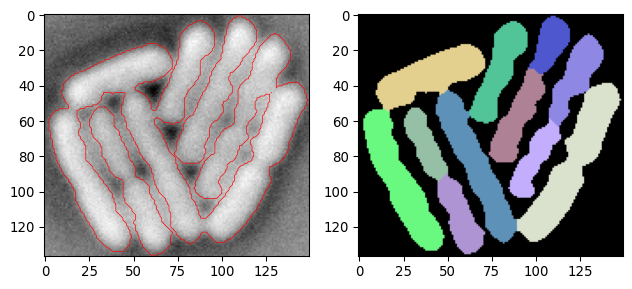
current_segmask, current_img_inv_crop = \
seg_bacterium(invert(imgs_ecoli[250,:,:]))
plot_img_and_segresult(current_img_inv_crop, current_segmask)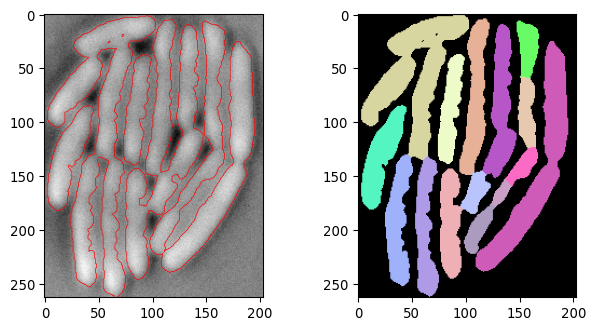
current_segmask, current_img_inv_crop = \
seg_bacterium(invert(imgs_ecoli[500,:,:]))
plot_img_and_segresult(current_img_inv_crop, current_segmask)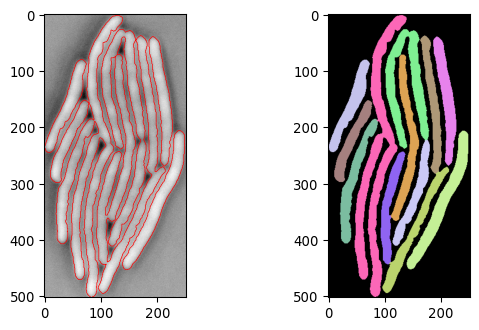
current_segmask, current_img_inv_crop = \
seg_bacterium(invert(imgs_ecoli[1000,:,:]))
plot_img_and_segresult(current_img_inv_crop, current_segmask)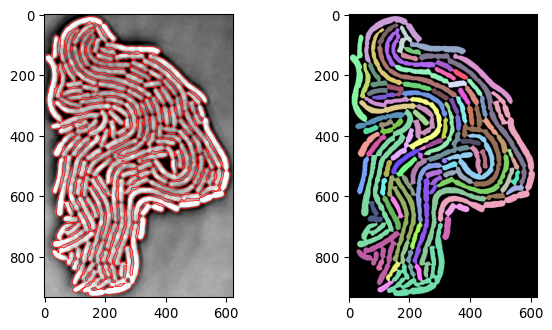
current_segmask, current_img_inv_crop = \
seg_bacterium(invert(imgs_ecoli[1050,:,:]))
plot_img_and_segresult(current_img_inv_crop, current_segmask)